Sarah Rauscher
email: sarah.rauscher@utoronto.ca
Education
2004 – 2011 PhD, Department of Biochemistry University of Toronto, Toronto, Ontario, Canada
2000 – 2004 Honours BSc, Major in Physics, Minor in Chemistry McMaster University, Hamilton, Ontario, Canada
Research Contributions
Book Chapter
Rauscher, S. and Pomès, R. (2011) Structural Disorder and Protein Elasticity. in Fuzziness: Structural Disorder in Protein Complexes, edited by Peter Tompa and Monika Fuxreiter (Landes Biosciences).
Peer-Reviewed Publications
- Li, G., Rauscher, S., Baud, S., and Pomès, R. (2011) Binding of Inositol Stereoisomers to Model Amyloidogenic Peptides. Journal of Physical Chemistry B [published online ASAP]
- Nikolic, A., Baud, S., Rauscher, S. and Pomès, R. (2011) Molecular mechanism of β-sheet self-organization at water-hydrophobic interfaces. Proteins 79, 1-22.
- Rauscher, S. and Pomès, R. (2010) Simulated Tempering Distributed Replica Sampling: A Practical Guide to Enhanced Conformational Sampling. Journal of Physics: Conference Series 256, 012011.
- Rauscher, S. and Pomès, R. (2010) Molecular Simulations of Protein Disorder. Biochemistry and Cell Biology 88, 269-290.
- Rauscher, S., Neale, C. and Pomès, R. (2009) Simulated Tempering Distributed Replica Sampling, Virtual Replica Exchange, and Other Generalized-Ensemble Methods for Conformational Sampling. Journal of Chemical Theory and Computation 5, 2640-2662.
- Rauscher, S., Baud, S., Miao, M., Keeley, F. W. and Pomès, R. (2006) Proline and Glycine control protein self-organization into elastomeric or amyloid fibrils. Structure 14, 1667-1676.
- Knapp, D., Kallin, C., Ghosal, A. and Mansour, S. (2005) Antiferromagnetism and charged vortices in high-Tc superconductors. Physical Review B 71, 064504.
- Cozzolino, A. F., Vargas-Baca, I. Mansour, S. and Mahmoudkhani, A. H. (2005) The Nature of the Supramolecular Association of 1,2,5-Chalcogenadiazoles. Journal of the American Chemical Society 9, 3184-3190.
- Zhang, W., Cozzolino, A. F., Mahmoudkhani, A. H., Tulumello, M., Mansour, S. and Vargas-Baca, I. (2005) Influence of π-Stacking on the Resonant Enhancement of the Second-Order Nonlinear Optical Response of Dipolar Chromophores. Journal of Physical Chemistry B 109, 18378-18384.
- Mahmoudkhani, A. H., Rauscher, S., Grajales, B. and Vargas-Baca, I. (2003) Structural Diversity of Lithium Sulfenamides: 7Li NMR Studies in Solution and Crystal Structures of [Li2(η2CH3)3C-NS-C6H4CH3-4)2(THF)2] and [Li2(η1-4-CH3C6H4-NS-C6H4CH3-4)2(THF)4]. Inorganic Chemistry 42, 3849-3855.
Conference Proceedings
Seo, M., Rauscher, S., Pomès, R., and Tieleman, D. P. (2011) Improving Internal Peptide Dynamics in the Coarse-Grained MARTINI Model: Application to Amyloid and Elastin Peptides. From Computational Biophysics to Systems Biology 2011, Jülich, Germany.
Current Research Interests
Molecular Simulations of Protein Disorder
Background
Disordered proteins are abundant in all kingdoms of life: more than one-third of mammalian proteins and more than 75% of cancer-associated proteins are predicted to contain disordered regions. Despite their biological importance, intrinsically disordered proteins (IDPs) are poorly understood relative to the wealth of structural data available for folded proteins in the Protein Data Bank. The bioinformatics of disordered proteins has advanced significantly in the past decade, and disordered regions can now be predicted with ~80% accuracy. While it is now possible to identify IDPs, they are notoriously difficult to study using experimental approaches. Their tendency to aggregate in solution presents significant challenges. However, more fundamentally, disordered proteins and disordered regions of proteins have many energetically-accessible conformational states (thousands or millions of states? – we don’t yet have an adequate description of their conformational landscape to describe the phenomenon quantitatively). Site-specific measurements obtained using NMR or fluorescence spectroscopy represent ensemble averages (population-weighted over many conformational states). Even with hundreds of experimental observables (for the best-characterized IDPs), it is not possible to obtain an unambiguously-determined ensemble of conformations. This is because the structural characterization of disordered proteins is an inherently under-determined problem: a small number of restraints are insufficient to uniquely define the conformations of a system with thousands of degrees of freedom. Molecular simulations, with their empirical force fields, can offer the additional information required to obtain conformational ensembles for disordered states of proteins. We have written a review discussing the application of molecular simulations to disordered states of proteins (see publication 4).
Rationale
The research goal of my present work is to obtain fundamental insight into the structural and physicochemical properties of disordered states of proteins using molecular simulation. While no experimental data for disordered proteins is directly incorporated into our models, we have, where possible, compared to experimental data to test the ability of empirical force fields to reproduce the essential structural features of these systems.
Enhanced Conformational Sampling
A key step in this study was to develop and test an enhanced sampling methodology (simulated tempering distributed replica sampling – STDR). The STDR algorithm consists of an alternation of two steps: (1) a short MD simulation (a few picoseconds in length), and (2) a Monte Carlo move in temperature. Repeating these two steps thousands of times results in a random walk in temperature. Performing simulations that employ a random walk in temperature is an ideal way to cross over barriers in the energy landscape and sample all relevant conformations. This is analogous to the commonly-used replica exchange method, but in practice offers enhanced computational efficiency and numerous practical advantages. Unlike replica exchange, STDR does not require replicas to perform synchronized temperature exchanges, and requires minimal communication between replicas. Importantly, STDR is well-suited for shared computing platforms because readily it accommodates fluctuations in CPU availability. The simulations in this study are both computationally demanding and data intensive: more than 50 million CPU hours on the SciNet supercomputer were required, resulting in more than 45 TB of data. In-house software was developed to perform both the STDR simulations and the analysis. The details of the STDR algorithm and implementation are described in publications 3 and 5.
Elastin
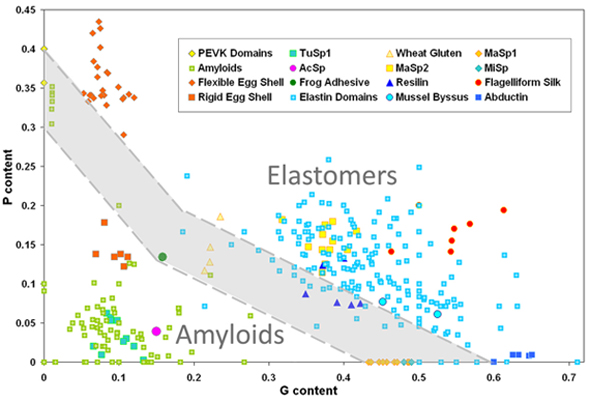
Elastin is an intrinsically disordered protein that confers the propensity for elastic recoil on biological tissues, including lungs, large arteries, and skin. Its rubber-like elasticity and remarkable elastic mechanical properties (high resilience, low elastic modulus) are due to its structural disorder. Thermoelasticity measurements have shown that the driving force for elastic recoil is primarily due to the higher entropy of the relaxed state relative to the stretched state. Thus, elastin performs its biological function by virtue of its disorder. Elastin is actually part of a larger class of proteins that exhibit rubber-like elasticity, which also includes resilin, abductin, colP from mussel byssus threads, and flagelliform and major ampullate spider silk. We have written a review that discusses structural disorder in the context of rubber-like elastomeric proteins (refer to our book chapter). We have also shown in an earlier study (publication 6) that elastin-like and amyloidogenic sequences are separable on the basis of hydration and conformational disorder, and that these properties are modulated by the amino acids proline and glycine. The generalization of this finding to the structure and function of elastomeric proteins was confirmed by our observation of an approximate threshold in combined proline and glycine content separating the composition of known amyloidogenic sequences from those of known elastomeric proteins (Figure 1). Remarkably, the compliance with a PG composition threshold is not limited to the hydrophobic domains of elastin, but is also observed for the elastic domains of ColP, abductin, resilin and spider silk.
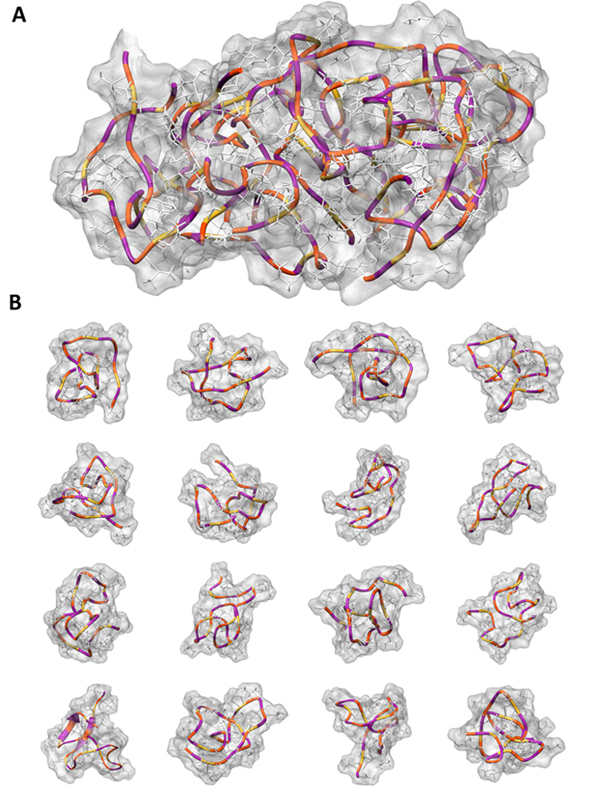
We have performed extensive enhanced sampling simulations of the peptide (GVPGV)7, which is an elastin-like motif from exon 24 of chicken elastin. We have obtained an atomic-level description of the disordered conformational ensemble of this peptide (Figure 2) as both a monomer (B) and an aggregate with 8 peptides (A). A selection of conformations from the simulation is shown, which represent a very small subset of the thousands of possible conformations of this peptide. These simulations push the boundaries of modern atomistic molecular dynamics simulations, requiring 84 μs for the monomer and 153 μs for the aggregate. A detailed analysis of the conformations from these simulations is ongoing.