

Faculty Listing
In
Vitro Protein Folding |
Molecular Chaperones |
Computational Protein Folding |
Diseases
of Misfolding and Aggregation |
|
Chalikian |
Chan |
Adeli |
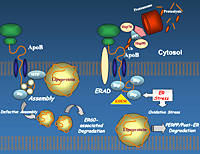 |
Our laboratory is studying the links between ER stress and the folding, ER translocation, and intracellular degradation of apolipoprotein B (apoB), a very large 4536 amino acid protein expressed in hepatocytes. ApoB is a very important protein in regulating human cholesterol and fat metabolism, and its hepatic production is increased in patients with diabetes and cardiovascular disease (thus the interest in studying mechanisms regulating its biosynthesis). ApoB is a highly hydrophobic protein that requires co-translational lipidation for correct folding of the emerging nascent chain in the ER. Impaired ER translocation or folding of nascent apoB (e.g. in the absence of sufficient lipid supply) leads to accumulation of the protein in the ER and rapid degradation. We plan to modulate the lipidation and folding state of nascent apoB chains and determine the upstream effects on ER translocation and apoB mRNA translational efficiency. Based on recent preliminary data (suggesting that misfolded apoB itself induces ER stress), we also plan to determine the potential role of misfolded nascent apoB in inducing ER stress via PERK signaling. Accumulation of folding-incompetent proteins in the ER can normally disrupt ER and activate the unfolded protein response (UPR). UPR activates an ER resident transmembrane protein: double-stranded RNA-activated protein kinase (PKR)-like ER kinase (PERK), which phophorylates eIF2alpha. Collectively, these studies will help us delineate the links between apoB misfolding, PERK signaling and upstream translational control of apoB mRNA. Website: http://www.sickkids.ca/Research/Adeli-Lab/index.html |
 |
In Alzheimer's disease, insoluble fibrils composed of Ab peptides accumulate to form senile plaques. A long-standing hypothesis has been that these fibrils are neurotoxic and cause Alzheimer's disease. Recently, however, a non-fibrillar soluble oligomer of Ab has been found to inhibit long-term potentiation, a process that is critical for learning and memory. These findings implicate soluble Ab oligomers in the pathogenesis of Alzheimer's disease. We have found that soluble Ab oligomers can be formed and stabilized for extended periods of time. Through kinetic experiments we are uncovering how the oligomers form and then further assemble into more complicated structures that culminate in amyloid fibril formation. Current projects include the following: (a) to use sedimentation methods to determine size of Ab oligomers, (b) characterize the structure of Ab oligomers using biophysical techniques, (c) determine whether Ab oligomers form under physiological conditions, (d) determine whether Ab oligomers are toxic to neurons and neuron-like cells. Website: http://www.oci.utoronto.ca/institutes/html/oci/msb/chakrabartty.html |
 |
Understanding molecular forces, including hydration, that stabilize various protein conformations is vital for our ability to develop strategies for tackling the protein folding problem. One approach is to combine thermodynamic and structural properties of proteins in an attempt to identify and thermodynamically characterize a variety of interactions that stabilize/destabilize a given protein state. In line with this strategy, we use high precision densimetric, ultrasonic velocimetric, calorimetric, and spectroscopic techniques to characterize the initinsic and hydration properties of a startegically selected set of globular proteins in their native and denatured states. In these studies, which represent the initial step of our research program, we investigate structurally well characterized, relatively small globular proteins. As the work progresses, we gradually expand our experimental targets to additional proteins to fully exploit the powers that the combination of volumetric, calorimetric, and spectroscopic techniques provides for understanding the role of individual molecular interactions in protein folding. Website: http://www.pharmacy.utoronto.ca/graduate/faculty/chalik.jsp |
 |
A hierarchy of heteropolymer models of various complexities are being developed to capture the essential physics of protein folding. These constructs allow for a broad coverage of conformational space -- in some cases also sequence space -- at a level of relative mathematical rigor currently not achievable in higher-resolution protein models. Physical insights are being gained into general features of protein folding, including calorimetric two-state cooperativity, chevron plots, and the effects of temperature and denaturant dependencies of hydrophobic interactions on native stability and folding/unfolding kinetics. The polymer-physics-based sequence-structure mappings afforded by some of these models are also applied to theories of evolutionary landscapes. A closely related research area is statistical mechanical theories and atomic simulations of aqueous solvation. This effort aims to attain a fundamental understanding of hydrophobic and other solvent-mediated interactions by simulating potentials of mean force of model-compound solutes in explicit water. Website: http://biochemistry.utoronto.ca/chan/bch.html |
 |
My laboratory is investigating the protein folding problem using both in vitro experimental and bioinformatic approaches. We are using the SH3 domain as a model system to address a variety of issues in the field of protein folding. SH3 domains are 60 residue protein-protein interaction modules, which are found in a wide variety of larger proteins involved in myriad intracellular signaling pathways. Our approach to studying protein folding is to perturb atomic interactions within the protein using mutagenesis, and then to characterize the changes in thermodynamic stability, protein folding kinetics, and peptide binding function caused by these perturbations using a variety of biophysical techniques. By the analysis of large numbers of mutants, we are able to illuminate the mechanisms by which proteins fold. We also use rigorous statistical analyses of protein sequence alignments to aid in interpreting our experimental results, and in predicting the effects of amino acid substitutions. Website: http://lambda.med.utoronto.ca/Davidson/ |
 |
Genetic mutations in the transmembrane (TM) helices of proteins underlie the causes of many human diseases, including several forms of cancer, diabetes, cystic fibrosis, and muscular dystrophy. These domains have been characterized in our lab by several criteria, including composition, sequence, and hydrophobicity. Using this background knowedge, we are now focusing on protein folding as follows: (i) Bioinformatics approaches to disease-causing mutations in membrane proteins - As polar TM mutations produce a striking variety of genetic diseases, we are determining the 'phenotypic propensities' and scope of mutation patterns of both polar and non-polar residues in TM domains, in conjunction with how inter-helical polar side chain-polar side chain hydrogen bond formation/loss impacts on protein folding within membranes. (ii) Hydrophobic patches in soluble proteins as nucleation sites for folding - Evidence from the hydrophobicity algorithm TM Finder suggests that short hydrophobic sequences in soluble proteins are membrane-insertion competent yet too short to act as TM segments. We are investigating proteins with two or more such patches to determine their role in guiding tertiary structure development. Website: http://www.sickkids.ca/aboutsickkids/directory/people/d/charles-deber.html |
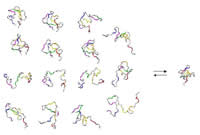 |
For a full understanding of protein folding and stability, characterization of unfolded and other disordered states of proteins is required. In addition, studies of disordered states are critical in order to probe the range of biologically relevant conformations and their roles in protein recognition since chaperones, transport proteins and proteolysis machinery recognize disordered states and many proteins are unstructured under native conditions, particularly in the absense of binding targets. Our work is focussed on the characterization of disordered states of protein using nuclear magnetic resonance (NMR), other spectroscopic and computational approaches. Our goals are to characterize preferential structure in disordered states, measure kinetic and thermodynamic properties, calculate conformational ensembles of disordered states and perform computational simulations. In addition, we are investigating the structures and dynamics of protein chaperones and their interactions with disordered proteins. Website: http://www.sickkids.ca/aboutsickkids/directory/people/f/julie-forman-kay-staff-profile.html |
 |
Alzheimer Amyloid
- Structure and Function Presenilins
- Biochemistry and Structure Website: http://medbio.utoronto.ca/faculty/fraser.html |
 |
In the Glover lab, yeast is used as a model system to examine the roles of molecular chaperones in protein folding in different cellular compartments. One focus of the lab is the oligomeric ATPase Hsp104. The unique ability of Hsp104 remodel aggregated proteins is thought to be the basis of its vital contribution to thermotolerance in yeast and to the stability of various yeast prions- novel protein-based genetic elements. The current research on Hsp104 involves establishing the key features of Hsp104-mediated protein extraction from aggregates and its interactions with yeast prion proteins. The Glover lab is also studying the chaperones in the yeast endoplasmic reticulum. Perturbation of protein folding homeostasis in the ER elicits a specific stress response. Chronic ER stress is thought to lead to cardiovascular complications in some types diabetes and the genetic disorder hyperhomocysteinemia in humans. The role of chaperones in maintenance of ER function is being investigated. Website: http://biochemistry.utoronto.ca/glover/lab/ |
 |
The Gradinaru biophysics laboratory employs advanced laser and detection technology to capture structural and functional dynamics of individual biomolecules. Single-molecule techniques are widely applied to the study of protein folding, enzymes, chemical receptors and biosensors, due to a unique capability to "see" beyond the intrinsic disorder and complexity of biological systems and to better connect with theory. The group has designed and built fluorescence microscopes capable of simultaneous recording of multiple properties of weak optical signals emitted by single probes. Various spectroscopy and imaging capabilities are available in the lab, such as multiparameter single-molecule fluorescence, burst, FRET, FRAP, FCS, PCH and multichannel WF/TIRF microscopy. Currently, we are focused on the study of rapidly fluctuating structures of intrinsically disordered protein and of the inhibitory peptidic drugs against the transcription protein Stat3. We are also investigating the structure of thermosensitive lipid-coated polymeric beads for drug delivery and the crowding-induced anomalous diffusion of proteins in the nucleus of live cells. Website: http://www.utm.utoronto.ca/~w3gradin/ |
 |
The ultimate aim of our projects is to address the fundamental question of how molecular chaperones modulate protein folding in the cell. To this end, we study the role of molecular chaperones in maintaining protein homeostasis using E. coli and yeast as model systems. We are also mapping what we call the chaperone interaction networks in yeast with the ultimate aim of identifying the rules that govern protein folding processes in the cell. Our group employs a battery of approaches including biochemical, biophysical, proteomics, and bioinformatics tools. The two chaperone families that we are concentrated on include the Hsp100 family and the Hsp90. Website:
http://biochemistry.utoronto.ca/houry/Labpage |
 |
My laboratory is interested in elucidating structure, dynamics, and interaction of biological macromolecules in order to understand, at the molecular and structural level, how they function in the cell. Of particular interest are proteins involved in calcium signaling, cell-cell interactions and transcriptional regulation. Our recent studies highlighted a significance of protein folding events in those cellular functions. For example, our studies on the calcium signaling protein STIM1 (stromal interaction molecule 1) revealed that the activation of STIM1-mediated store-operated calcium entry (SOCE) employs a molecular mechanism in which protein unfolding coupled with oligomerization process plays a crucial role. STIM1 and its target calcium channel termed Orai1 are both implicated in genetic diseases such as severe-combined immune disease (SCID). Our ultimate goal is to find ways to cure this and related human diseases including cancer. Our primary tool is NMR spectroscopy, but we also use a variety of biophysical and molecular biology techniques, including X-ray crystallography and in vivo fluorescence imaging microscopy. Website: http://nmr.uhnres.utoronto.ca/ikura |
 |
NMR methods are developed and applied to study a wide range of biophysical problems. In the area of protein folding and molecular dynamics new triple resonance NMR experiments are designed to probe residual structure, dynamics and thermodynamics of unfolded and partially unfolded states. Relationships between dynamics and thermodynamics are being developed and applied to unfolding events. Website: http://abragam.med.utoronto.ca/UTNMR.html |
 |
Our studies related to protein folding fall into two categories. Firstly, the
sulfoglycolipid (SGL) binding of the N-terminal ATPase containing
domain of hsp70. We have shown that SGL binding to this domain inhibits
hsp70 ATPase activity and are currently investigating the effect
of this binding on hsp70 chaperone function in several assays: a)
SV40 The second
area involves the intracellular trafficking of CFTR and how this
relates to Website: http://www.sickkids.ca/Research/Lingwood-lab/index.html |
 |
Extra- and intra- cellular macromolecules, many of which contain carbohydrates, are degraded in a stepwise manner in the lysosome and their components subsequently recycled. Specific lysosomal exoglycosidases are involved in this process. If one of these enzymes is deficient, partially degraded macromolecules are trapped in the lysosomes, resulting in disease. Together, lysosomal storage diseases (LSDs) occur at a rate of ~1 in 5000 births, making them a significant health burden. Like secretory proteins, lysosomal enzymes are synthesized in the endoplasmic reticulum (ER). Once they fold and pass the ER’s quality control system they are transported through the Golgi and targeted to lysosomes. Many LSDs are caused by the inability of the newly synthesized mutant protein to fold or remain folded in the ER, leading to enzyme deficiencies and substrate storage. We postulate that clinical symptoms are exacerbated by the presence of the unfolded mutant protein itself, which initiates the unfolded protein response (UPR) leading to ER-stress, and cell death. Currently there are two major approaches to treat these disease, enzyme replacement (ERT) and substrate reduction (SRT) therapy. The former costs ~$300,000/ yr./ patient, which limits availability, and the only compound presently being used for the latter has unpleasant side effects that limit its dosage/ efficacy. Neither of these approaches address the problems caused by the UPR. Additionally, ERT is unable to deliver enzyme to the CNS, or other organ compartments. We are developing enzyme enhancement therapy (EET) as a new therapeutic approach. EET uses small molecules that bind to and stabilize the properly folded form of the enzyme, overcoming the destabilizing effects of the mutation. EET-agents are known as chemical or pharmacological chaperones and often are competitive inhibitors of the target enzyme. Additionally we are investigating the pathways and mechanisms that mutations in selected LSDs initiate to produce the UPR in cells. Website: http://www.sickkids.ca/aboutsickkids/directory/people/m/don-mahuran-staff-profile.html |
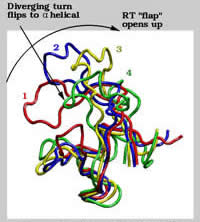 |
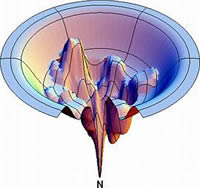
We use structural models derived from spectroscopic studies together with fundamental physical laws governing atomic interactions to simulate the time evolution of biomolecular systems. With modern supercomputers, these molecular "cartoons" typically cover time scales of 10-15 to 10-8 s, a time span relevant to such important biophysical processes as enzymatic reactions, molecular recognition and binding, ion transport across biomembranes, and the proper folding and function of proteins. The atomic resolution and the wealth of details provided by the simulations make them a useful tool to complement experimental studies. In collaboration with other research groups in Toronto and elsewhere, we study equilibrium properties and dynamic fluctuations involved in the function of soluble and membrane proteins, in folding-unfolding processes, and in protein-ligand association. The attached figure depicts collective motions in thermally-induced unfolding of the N-terminal SH3 domain from drk. Website:
http://www.pomeslab.com/
|
 |
We are studying the fundamental properties of protein-lipid and protein detergent interactions with an emphasis on the thermodynamics of membrane protein folding. We are particularly interested in characterizing the detergent-solubilized state of membrane proteins. This is important for the structural study of membrane proteins by crystallography and NMR, but this also gives us insight into the intrinsic properties of integral membrane proteins. We have developed a method to refold protein from the SDS-denatured state by the addition of certain small amphipathic alcohols, such a 2-methyl-2,4-pentanediol. We are currently trying to understand the physical basis for this effect. We are looking the interactions of model peptides with a series of detergents and cosolvents with calorimetric, spectroscopic and molecular dynamic methods. We are also developing lipopeptide detergents (LPDs), a fundamentally different class of amphiphile designed to better mimic lipid bilayers. LPDs result in dramatic increases in the thermostability of solubilized membrane proteins relative to traditional detergents. We use de novo principles in designing our LPD peptide sequences and structures. Website:
http://xtal.uhnres.utoronto.ca/prive |
 |
We are interested in protein folding mechanisms through solution state NMR studies, of calmodulin, a key soluble protein involved in calcium mediated signaling. By NMR, it is possible to track residue-specific chemical shifts along the protein folding pathway and in so doing, reveal potential folding intermediates, cooperative processes, and regions where folding is initiated. We make use of additional biophysical techniques to better understand the folding mechanism. For example, by dissolving oxygen into the sample or by exploring solvent isotope effects we can “see” solvent exposed surface area for each residue, along the folding pathway. This helps us understand the sequence of events associated with folding events. We also routinely make use of pressure and biosynthetic labeling with 19F probes to gain insight into the nature of the unfolded states, via NMR. This approach can be extended to in vivo conditions, where viscosity and crowding, plus binding partners often cause linewidths to increase by a factor of 5 or more. Our solution is to “tag” target domains in protein samples with robust and well-resolved monomethyl groups such that NMR can be used to monitor folding events and post-translational modifications, upon microinjection or trafficking of labeled proteins into Xenopus oocytes or yeast cells. Website:
http://fourier.utm.utoronto.ca |
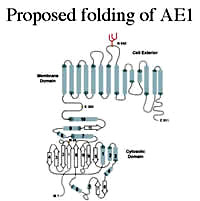 |
My laboratory is interested in the structure, function and assembly of membrane proteins, with a particular focus on the anion exchanger (AE1) found in erythrocytes and in acid-secreting intercalated cells in the kidney. This membrane glycoprotein catalyses the electro-neutral exchange of chloride and bicarbonate across plasma membranes and is involved in bicarbonate transport, intracellular pH regulation and acid secretion. Mutations in the AE1 gene cause a number of human diseases, including Southeast Asian ovalocytosis (SAO) and hereditary spherocytosis (HS) that affect red blood cells, and distal renal tubular acidosis (dRTA) that affects acid secretion by the kidney. In an extensive set of studies using transfected cells we have shown that the mutations cause mis-folding of AE1 and impaired trafficking to the cell surface. We are studying the interaction of chaperones and other components of the quality control machinery of the cell with normal and mutant forms of AE1 in order to determine their role in the retention of AE1 mutants. High-throughput analyses (membrane yeast 2-hybrid and proteomics) are being used to identify the full complement of cellular proteins that interact with normal and mutant forms of AE1. We are also carrying out biophysical and NMR studies to determine the effect of mutations on the structure and stability of the cytosolic domain of AE1 expressed in E. coli. The overall goal is to determine the effect of mutations linked to disease on the folding and trafficking of AE1 and the cellular components involved in this process. Website:
http://reithmeier-lab03.med.utoronto.ca/~reinhartreithmeier/lab/ |
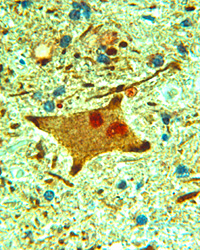 |
Amyotrophic lateral sclerosis, or Lou Gehrig’s disease, is characterized by the presence of proteinaceous aggregates in motor neurons of the brain and spinal cord, the neuronal type affected in the disease. Our aim is to understand how these aggregates are formed and to establish their relationship to the neurodegenerative mechanism. The two major proteins of interest in our laboratory are the neuronal intermediate filament protein, peripherin, and superoxide dismutase-1 (SOD1). Mutations within SOD1 account for ~3% of all ALS cases and the mutant SOD1 protein has a propensity to misfold and to aggregate. Peripherin aggregates are found in 80% of all ALS cases and transgenic mice overexpressing peripherin develop a motor neuron disease. We use molecular cell biology approaches, including primary cell culture, transgenic mouse models of ALS and human pathological tissue, together with protein-biochemical techniques to understand how a protein aggregate leads to neuronal death. Image: Motor neuron in ALS spinal cord double-labeled with antibody to peripherin (brown) and ubiquitin (red). Ubiquitin labels various structures within diseased motor neurons, appearing as aggregated material. Section is counterstained with haemotoxylin and eosin. Normal control tissue is negative for peripherin and ubiquitin immunoreactivity. X40 magnification. Website:
http://icarus.med.utoronto.ca/patho/faculty.asp?FacultyID=425 |
 |
Our work contributes
to two strands of research at the interface of proteomics and neurodegenerative
disease research: the development of strategies for the study of
protein interactions and the application of these strategies to
address questions in the context of neurodegenerative disease research. Website: http://individual.utoronto.ca/cpu/index.htm |
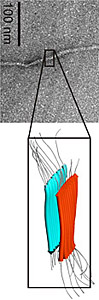 |
Our research focuses on elucidating the structure and mechanisms of assembly of macromolecular peptide/ protein complexes, with a particular emphasis on membrane-active proteins involved in human disease. Specific systems being investigated in the lab include: (1) Non-fibrillar oligomers formed by amyloid peptides. These have been implicated as the key cytotoxic agents in numerous neurodegenerative diseases, and we are working to define the molecular mechanism of their activity at cell membranes. (2) Early misfolding intermediates formed by the mammalian prion protein (PrP). Conversion of PrP from a native helical form to the misfolded, beta-sheet rich scrapie form is the hallmark of prion diseases, such as Creutzfeldt-Jakob disease or bovine spongiform encephalopathies. Understanding the structure of oligomeric species formed early in the misfolding pathway will shed light on the mechanisms of conversion and disease progression. (3) Viral pore/channel proteins (viroporins). These proteins play an important role in the life cycle of several viral pathogens, including HIV-1, hepatitis C and dengue virus. We are studying the structure and functional assembly of these proteins as a first step towards development of new antiviral therapies. Website: http://www.sickkids.ca/Research/Sharpe-lab/index.html |
|
Website: http://biochemistry.utoronto.ca/steipe/ |
|
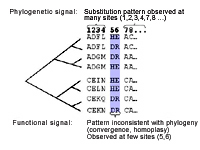 |
My lab is currently working on the development of computational methods for the analysis of protein sequences. Sequence information is exploding such that a large sampling of the possible functional sequence space that is explored through evolution is available for analysis. Analyses of sequence alignments from protein families contain much information on their function, structure and evolution. The development of accurate analysis tools permits the prediction of amino acids involved in protein folding, structure, function and even protein-protein interaction. Current analyses rely on absolute conservation of amino acid sites, where the methods we are developing also consider co-variation patterns between amino acid sites within and between proteins. Analyses of specific proteins of interest will allow us to infer the important residues involved in protein structure and function and give clues on the nature of disease involving these proteins and the potential for designed targeting of particular residues by drugs. Additionally, large scale high-throuput analyses of gene families may allow the discovery of general biochemical principles of protein function. We have also been working in collaboration with Walid Houry (Biochemistry, University of Toronto) in a study of the phylogenetic distribution of Molecular Chaperones. The impetus of this work has been the conspicuous absence of the GroEL chaperone in several sequenced genomes of Mycoplasma. Using comparative genomic analyses we are investigating mechanisms by which the bacteria can cope with the loss of such an important protein folding chaperone. Website: http://www.uhnres.utoronto.ca/tillier/ |
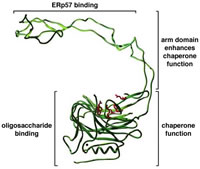 |
Protein folding in vivo differs from the situation in a test tube in that nascent polypeptides attempt to fold even before their entire length has been synthesized. This should lead to aggregation. However, in vivo, molecular chaperones recognize hydrophobic segments of non-native polypeptides and, through regulated cycles of binding and release, prevent aggregation and allow productive folding to occur. We study protein folding within the the endoplasmic reticulum, a unique environment wherein polypeptides are glycosylated, acquire disulfide bonds, and undergo folding. Our emphasis is on the chaperones calnexin and calreticulin; truly remarkable proteins that assist folding by binding both to unfolded polypeptide segments and to oligosaccharide chains. They also assist folding by recruiting a thiol oxidoreductase to the unfolded substrate. To determine how these diverse functions are organized and regulated, we use a combination of approaches that include monitoring of folding in vivo, structure-function mutagenesis studies, detection of novel folding factors, and reconstituting folding in vitro with defined components. Website: http://biochem17.med.utoronto.ca/%7Edavid_williams/lab |
|
My protein
folding related research includes the following topics: Website: http://biochemistry.utoronto.ca/wodak/bch.html |
|
 |
Our research
is aimed at finding general strategies for the photo-control of
peptide and protein structure – and thereby activity. In doing
so, we gain fundamental knowledge about protein folding, and dynamics.
We also develop powerful tools for the analysis of complex biochemical
systems. Website: http://www.chem.utoronto.ca/staff/GAW/ |
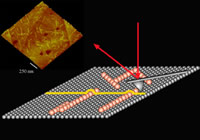 |
Our group is fundamentally interested in understanding how biomolecules self-assemble hence our focus on single molecule biophysics. Self-assembly is the formation of complex hierarchical structures from their individual atomic or molecular elements without external intervention. By teasing out key structural and chemical parameters, which often hinges on detailed characterization of the interface between the constituent molecules or aggregates, we can develop a set of "rules" or precepts critical to efforts on molecular and protein engineering. To study these mechanisms over all dimensional scales from individual molecular complexes to the formation of two and three-dimensional supramolecular assemblies, we are combining traditional approaches such as circular dichroism, light scattering, and spectroscopy, with high-resolution in situ scanning probe microscopy (SPM). Our research programs also include computational approaches to simulating protein assembly and a strong effort into the design, fabrication, and application of functional single molecule imaging tools. Website:
http://bigten.med.utoronto.ca |
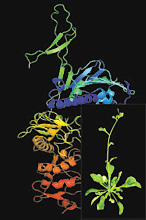 |
Organisms such as plants do not have the ability to move and escape from environmental stresses and are more frequently affected adversely. Naturally, plants like most other organisms develop complex mechanisms to sensor and cope with environmental stresses in which processes the normal protein folding and function are challenged. Our interests are to understand how molecular chaperones, most of which are highly induced to help the other proteins to function properly under stress conditions, are involved in plant stress resistance and in the process of normal plant development. Currently, the research is focused on the function and the mechanism of action of molecular chaperone Hsp90 family in the model plant Arabidopsis thaliana. On the other hand, we are also interested in the role of selective protein degradation by the proteasome in the control of cellular protein homeostasis. By using yeast Saccharomyces cerevisiae as a model organism, we are interested in studying how molecular chaperones are involved in de novo protein synthesis and the interplay of chaperones and the proteasome in the cellular protein quality control systems upon protein misfolding by means of biochemical, biophysical and proteomics approaches. Website:
http://www.csb.utoronto.ca/faculty/zhao-rongmin
|
| Home | Program Description | Faculty | Trainees | How to Apply | Events and Information | Publications | Copyright © 2002. CIHR Training Program in Protein Folding |